
Abstract
Knowing which drivers affect the spatial distribution of hybridizing species and their admixed individuals on local or regional scale can leverage our understanding about processes that shape taxonomic diversity. Hybridizing white oak species (Quercus sect. Quercus) represent an ideal study system to elucidate which environmental factors determine their relative abundance and admixture levels within admixed forest stands. To elaborate these relationships, we used 58 species-diagnostic single-nucleotide polymorphism (SNP) markers and high-resolution topographic and soil data to identify the environmental factors associated with taxonomic composition of individuals and populations in 15 mixed stands of Q. petraea and Q. pubescens in the Valais, an inner-Alpine valley in Switzerland. At the individual tree level, generalized linear models (GLMs) explained a relatively small part of variation (R2 = 0.32). At the population level, GLMs often explained a large part of variation (R2 = 0.54–0.69) of the taxonomic indices. Mean taxonomic composition of the sites depended mainly on altitude and geographic position. Moreover, the more within-site variation we found in predictors related to topographic position, the higher was the average genetic admixture of single trees. Our results show that a multitude of topographic and edaphic factors affect the taxonomic composition and admixture levels of these two hybridizing oak species on local scale and that regional heterogeneity of these factors promote taxonomic diversity and admixture. Overall, our study highlights the prospects of using tailored genetic resources and high-resolution environmental data to understand and predict taxonomic composition in response to changing environments.
Similar content being viewed by others

Introduction
A central question in evolutionary ecology is which environmental factors drive the spatial distribution of species. Ecological niche models (ENMs), also called species distribution models (SDMs), have traditionally been used to address this issue and have given insights into the ecological and evolutionary processes that determine species distribution on mostly large spatial scale (reviewed in, e.g., Elith and Leathwick 2009; Guisan et al. 2017). However, much less is known about what influences the spatial distribution of hybridizing species and their inherent genetic admixture in taxonomically mixed populations on small spatial scale. Such information would be highly valuable to conserve and promote biodiversity on regional and local scale and to study adaptive introgression (Suarez-Gonzalez et al. 2018).
Classically, hybrid complexes have been described as systems in which the two parental species persist along spatial and/or environmental gradients, with a hybrid zone in-between where hybrids are constantly produced and may even have a fitness advantage compared to their parental taxa (Abbott 2017). For example, the two parental species carrion and hooded crows (Corvus corone and C. cornix) have clear distributions in Eastern and Western Europe, respectively, and co-occur in a narrow range in Central Europe (Vijay et al. 2016), building a hybrid zone that is maintained by assortative mating and social marginalization (Randler 2007). In forest trees, such hybrid zones can be found over short or long spatial and environmental gradients like altitude, latitude, or soil moisture, for example, in the genera Abies, Picea, Populus, and Quercus (Abbott 2017). For instance, species integrity in Sitka, white, and Engelmann spruce (Picea spp.) in British Columbia is likely maintained by environmental selection in parental habitats, but hybridization and introgression can be found along fine-scale environmental gradients (Hamilton et al. 2015). In some cases, parental species and their hybrids can also occur in local sympatry. A classic example is the species complex of Louisiana Irises (Iris spp.), where species have clear ecological niches, but can be found, together with their hybrids, in sympatry within only a few meters (Cruzan and Arnold 1993). It has been shown that the spatial arrangement of the different taxa is non-random but, among others, associated to micro-environmental conditions and assortative mating (Cruzan and Arnold 1993; Johnston et al. 2001).
To answer the question which environmental factors are driving taxonomic composition and admixture within and between closely located populations of hybridizing species, one ideally studies a set of natural populations that (a) show high taxonomic variation (characterized by accurate species-diagnostic genetic markers), (b) span a large environmental gradient (described by high-resolution environmental data), but (c) are closely situated to exhibit historical and contemporary gene flow and admixture. European white oaks (Quercus sect. Quercus), in particular Quercus petraea, Q. pubescens, and Q. robur, represent an ideal study system to tackle this question. These species differ in their habitat requirements (e.g., Rellstab et al. 2016b), co-occur in mixed stands (e.g., Lepais and Gerber 2011; Rellstab et al. 2016a; Truffaut et al. 2017), and are known to hybridize and back-cross (e.g., Curtu et al. 2007a; Lepais and Gerber 2011; Leroy et al. 2017).
Various studies exist on the taxonomic composition, and admixture of white oak stands on local and regional scale. For example, using paternity analysis, Curtu et al. (2009) studied hybridization and introgression in a Romanian mixed forest stand containing four white oak species (Q. frainetto, Q. petraea, Q. pubescens, and Q. robur) and found that asymmetric gene flow due to reproductive barriers and relative abundance of the parental species has led to a mosaic of oak taxa on fine spatial scale. Lepais and Gerber (2011) investigated hybridization and introgression in a mixed stand of four white oak species (Q. petraea, Q. pubescens, Q. pyrenaica, and Q. robur) in southwestern France and discovered that species integrity was retained despite interspecific gene flow, but also detected asymmetric introgression by unequal contributions from pollen donors. Truffaut et al. (2017) used demographic monitoring and genetic methods to study adult and juvenile trees in one large mixed stand of Q. petraea and Q. robur in western France and showed ongoing hybridization and succession processes (from Q. robur to Q. petraea) on a fine spatial scale. All of these studies have not directly assessed the association of taxonomic composition and levels of admixture with environmental (micro-)conditions as potential drivers. More recently, Fu et al. (2022), using genome wide data, found that adaptive introgression between two widespread Asian non-white oak species, Quercus acutissima and Q. variabilis, was, among others, driven by environmental factors.
Species identification based on leaf morphological characters is notoriously difficult in white oak species, especially in Q. petraea and Q. pubescens where hybridization is abundant and leaf morphological characters can be highly overlapping (e.g., Bruschi et al. 2000; Rellstab et al. 2016a; Viscosi et al. 2009). Consequently, several sets of molecular markers have been developed to distinguish white oak species and classify admixed individuals (e.g.,Guichoux et al. 2011; Kampfer et al. 1998; Steinkellner et al. 1997). However, especially with microsatellite markers, the assignment of and differentiation between Q. petraea and Q. pubescens remains difficult (e.g., Curtu et al. 2007b; Neophytou 2014; Rellstab et al. 2016a). These two species exhibit a short phylogenetic distance in combination with extensive contemporary gene flow following recent secondary contact after a period of isolation (Bruschi et al. 2000; Leroy et al. 2017). However, the recently developed set of species-discriminatory single-nucleotide polymorphisms (SNPs) of Reutimann et al. (2020), based on reference individuals from across the European continent, has substantially improved species assignment, leading the way to accurate estimates of admixture levels.
Here, we used these species-diagnostic SNPs (Reutimann et al. 2020) together with high-resolution topographic and soil data to identify the environmental factors associated with individual taxonomic assignment, population-specific taxonomic composition, variation in taxonomic composition, and levels of admixture in 15 mixed stands of Q. petraea and Q. pubescens. The study took place in an area of about 7 km × 30 km in the Valais, the inner-Alpine upper Rhone valley in Switzerland. This valley is characterized by steep and rugged terrain that offers large gradients in topography and soil characteristics. It has been previously shown that Q. petraea and Q. pubescens co-occur and are admixed in this region (Gugerli et al. 2008; Rellstab et al. 2016a; Reutimann et al. 2020). Specifically, we addressed the following questions: (1) Do microclimatic environmental conditions allow to predict the taxonomic assignment of single trees? (2) Do environmental variables on regional scale (i.e., at population level) predict the taxonomic composition of populations? (3) Does environmental variation within population influence the taxonomic variation within the population? (4) Does environmental variation within population influence the admixture levels within the population?
Materials and methods
Study system
The two European white oak species Q. petraea (sessile oak) and Q. pubescens (pubescent oak) belong to the subgenus Quercus and are monoecious trees that occur over large areas throughout Europe. Sympatric white oak species have undergone extensive cytoplasmic exchange (Petit et al. 1993), and especially Q. petraea and Q. pubescens are known for elevated levels of interspecific gene flow (Gerber et al. 2014; Salvini et al. 2009). Despite little genetic and morphological differentiation (Bruschi et al. 2000; Rellstab et al. 2016a), the two parental taxa normally occupy slightly different, but overlapping habitat types. Quercus pubescens was shown to be more resistant to drought-induced air embolism than the more widespread and generalist Q. petraea (Cochard et al. 1992) and can survive extreme drought events (Galle et al. 2007). In addition, Q. pubescens generally occurs under warmer atmospheric temperatures and higher soil pH than Q. petraea (Leroy et al. 2020). Quercus robur, the third species included in the present dataset, but not a focus of the analyses, thrives on deep and moist soils and mainly occupies riparian hardwood forests. Regionally, it is mainly found in the westernmost part of the Valais, i.e., outside our study region.
The Valais, an inner alpine valley of Switzerland and the home of our study populations, provides suitable and overlapping habitats for both targeted white oak species and represents an ideal study area to investigate interspecific habitat differences on a regional scale. The Valais is characterized by steep slopes and high mountain ranges rising above 4000 m a.s.l., which shelter the inner part of the valley from the moist oceanic air masses transported with westerly and southerly winds. In the main valley, a dry-subcontinental climate prevails with high insolation, low precipitation (less than 600 mm precipitation per year), and an annual mean temperature of about 9.5 °C (period 1961–1990; MeteoSwiss). It is also characterized by high daily and seasonal temperature ranges, low humidity, and a strong precipitation gradient along the altitudinal gradient, which collectively are known to have shaped the composition of plant communities (Körner and Riedl 2021).
Sampling design
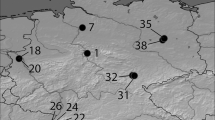
Our sampling design consisted of 15 closely located oak populations (238 individuals included in the final analysis) in the central Valais (Fig. 1, Table S1) between Grône and Visp, covering an area of around 210 km2 (7 km × 30 km). The distance between populations ranged from 1.4 to 31.6 km. Populations were selected from a larger set available from the Swiss Evoltree intensive study site (ISS, Lefèvre et al. 2016, https://www.evoltree.eu/resources/intensive-study-sites/sites/site/valais), maximizing the variability of topo-climatic conditions across populations. Six of the populations (87 trees) were genotyped and taxonomically assigned in Reutimann et al. (2020); the remaining nine populations (166 trees) were newly genotyped for this study. We assessed the geographic position of a reference point for each population with a hand-held GPS instrument (eTrex, Garmin, Schaffhausen, Switzerland). The precision of the localization is commonly at 2–5 m. From each reference point, we chose the closest, up to six adult oak trees along transects following the four main wind directions, leading to leaf samples from 10 to 23 trees per population for DNA extraction. The position of each tree was calculated by measuring distance and angle to the reference point with a laser-based Vertex instrument.

Sampling design and taxonomic characteristics of the 15 Quercus sect. Quercus populations. a Map of sampling locations represented as pie charts, which indicate mean genetically assigned taxon proportions (colors of charts) and admixture index (S, size of charts) averaged per population. Numbers correspond to simplified population labels. b Bar plot of all 238 sampled trees. Each vertical bar represents a single individual and colors reflect assignment probabilities to respective taxon clusters. Red lines show mean S (genetic admixture) at the population level. Populations are ordered from West to East
Environmental data
We derived topographic and edaphic data to characterize environmental conditions in each population at the stand and individual level. A previous study on a much larger spatial scale (Rellstab et al. 2016b) showed that soil water characteristics (described by topographic factors here), soil pH, and other topographic factors like slope differ among the habitats of white oak species in Switzerland. The original dataset consisted of geographic position, 14 topographic factors and one edaphic factor (Table S2), which were then reduced to 13 variables (Table 1, and see below).
For topography, we used a 2 m digital elevation model (swissALTI3D with a vertical precision of about ± 0.5 m; https://www.swisstopo.admin.ch/en/geodata/height/alti3d.html) to derive 14 variables (Table S2) at 4 m resolution based on biological hypotheses and their informative power on local scale (Leempoel et al. 2015). We generated morphometric, hydrologic, and radiation grids for Switzerland using SAGA 6.2 (Conrad et al. 2015) and extracted these variables with a bilinear interpolation method using the package raster v. 3.6.2 (Hijmans 2019) in R 4.0.2 (R Development Core Team 2020).
To include the edaphic dimension, we derived a topsoil pH layer for the forested area of Switzerland (Baltensweiler et al. 2021). This layer is based on 1944 forest soil profiles sampled by the Swiss Federal Research Institute WSL over the period from 1989 to 2018. We applied a random forest model implementation from the R-package caret v6.0–68 (Kuhn 2008), in which we used 80% of the soil profiles as training set and 20% as independent validation set. A total of 126 predictors were used as soil-forming factors to describe geology, topography, climate, and vegetation. The R2 of the validation set was 0.37, and the root mean square error was 1.08. The spatial resolution of the final pH layer is 25 m and is based on all soil profiles. Extraction was then carried out as described above.
The generalized linear models (GLMs) used below allow for n-2 predictor variables, where n is the number of sampling sites. Since we included 15 populations in this study, we reduced the set of 17 predictors described above to 13 predictors. Moreover, we wanted to exclude redundant information and limit collinearity in statistical models. To do so, we performed pairwise Pearson’s correlations between all environmental variables (Table S2) and applied the following rules to keep descriptors; (i) secondary (i.e., derived) variables that were highly correlated (r ≥|0.75|) with primary variables were removed (e.g., slope retained over morphometric protection index), and (ii) when two primary variables were highly correlated (r ≥|0.75|), we selected the one we considered biologically more relevant to test our hypotheses (e.g., direct solar radiation preferred over sky view factor). Based on this procedure, we retained 13 environmental variables for downstream analyses (Table 1, Table S2). All the retained variables were scaled and centered before performing the analyses.
Genotyping and taxonomic assignment
Extracted DNA was retrieved from the EvolTree DNA repository (Stierschneider et al. 2016). All samples were genotyped with a set of 58 unlinked, species-diagnostic SNPs described in Reutimann et al. (2020). These SNPs were shown to reliably discriminate between Q. petraea, Q. pubescens, and Q. robur and allow to estimate admixture levels of individuals and populations. Samples with more than 10% missing data were excluded from the analyses.
The taxonomic status of the genotyped individuals was assessed with the species assignment approach described in Reutimann et al. (2020). In brief, we used the training set (n = 194) developed in Reutimann et al. (2020) to assign the samples with Structure (Pritchard et al. 2000) using the USEPOPINFO parameter. When USEPOPINFO is turned on, information of individuals with POPFLAG = 1 (here training individuals) is used to group the remaining individuals. This ensures that individuals are assigned to predefined clusters and makes results more comparable and interpretable than without using the POPFLAG parameter. Structure was run with K = 3 (number of clusters, i.e., species), 10 iterations, and 1,000,000 repetitions after a burn-in period of 100,000 runs. For each individual, we obtained assignment probabilities to each of the three taxa included in the training, which are similar to co-ancestry coefficients, i.e., the proportion of the genome that derives from each of the three species. These assignment probabilities were used to calculate the Shannon’s diversity index (S) as an indicator of admixture within individuals. This index is scaled from zero to one, where a value of zero indicates no admixture and one means that an individual is fully admixed, i.e., it has equal assignment probabilities for all three species.
In the present study, we concentrated on assignment probabilities of Q. petraea and Q. pubescens. The third species, Q. robur, was only found in very low proportions (27 out of 238 trees show Q. robur probabilities of above 10% and maximal 30%; Fig. 1) and was thus not used as a main objective in the analyses. However, because they are part of the natural hybrid complex in white oaks, assignment probabilities of Q. robur were not removed from the dataset. Assignment probabilities of Q. petraea and Q. pubescens therefore do not necessarily sum up to 100%, and all analyses described below that use assignment probabilities as response variable were initially performed each for Q. petraea and Q. pubescens. Moreover, measures of admixture in individuals and populations, as well as of taxonomic variation of populations, also include Q. robur fractions.
Model selection
To address the specific questions that were introduced above and are described below in more detail, we applied two different model selection procedures, using the 13 retained explanatory variables (centered and scaled) and different response variables derived from genetically inferred assignment probabilities (Q values). We used the Q values of Q. pubescens as a basis for all the models, since it is the predominant species in the study area. The results using Q values of Q. petraea are highly similar and therefore not reported here. This is due to the fact that the Q values of the third species, Q. robur, are usually very low, leading to almost reciprocal Q values for Q. petraea.
Generalized linear models (GLMs) and generalized linear mixed models (GLMMs) were built with the gamlss function of the R-package gamlss (Rigby and Stasinopoulos 2005). The gamlss function is generally used for more complex generalized additive models; however, in this study, we used it for GLMs and GLMMs because of its flexibility to use different distribution families and link functions and to make the different model estimates better comparable with each other. A beta family with a logit link was used to model the distribution of response variables that are proportions derived from a continuous number (Douma and Weedon 2019) and a normal family with a logit link for normally distributed response variables such as variances. Random effects were added by specifying a random intercept model with the random function of gamlss. The best explaining models were identified using a stepwise model selection procedure in both directions (forward and backward) based on the generalized Akaike information criterion (GAIC) integrated in the stepGAIC function (default parameters, gamlss). To further minimize collinearity between predictor variables, a model resulting from the stepwise procedure that contained variables with a variance inflation factor (VIF) > 3 (Zuur et al. 2010) was run again, excluding the explanatory variable with the highest VIF as computed with the vif function of the R-package car (Fox and Weisberg 2019).
To further validate the relevance of the variables included in the final models after the stepwise model selection procedure, we applied Bayesian model averaging (BMA) with the bms function of the R-package BMS (Zeugner and Feldkircher 2015). In contrast to the GLMs/GLMMs described above, BMA does not identify a single best explaining model, but summarizes all possible (best) models and thereby provides additional information on the number, importance and sign of predictors. It uses Markov Chain Monte Carlo (MCMC) samplers to sort through model space and to assess the coefficients and posterior inclusion probability (PIP) of the covariates, posterior model size, and model probability (PMP). We performed BMA on models that contained the same covariates as the models before applying the stepwise model selection described above. The models were run with default settings, except that the length of burn-in was set to 1,000,000, number of iterations to 3,000,000, the number of best models to sample to 1000, and a uniform model prior was used. GLMMs could not be validated with this approach, as the inclusion of random effects is not possible with the BMS package. Posterior means of model coefficients produced by this procedure tend to underestimate the coefficients because they represent the coefficients averaged over all sampled models, including the model in which the variable was not included (Zeugner 2011). The values of posterior mean coefficients can therefore not be directly compared to coefficient estimates resulting from the stepwise model selection procedure, but the sign of the coefficients can be validated.
We implemented the following models to address our specific questions:
-
Question 1: Do micro-environmental conditions predict the taxonomic status of single trees? To answer this question, we used the 13 environmental descriptors (Table 1) to predict the taxonomic status of single trees (Q values of Q. pubescens). The first statistical model represents a GLM produced by the stepwise selection procedure that ignores site-level effects:
-
Model 1a: Q value ~ environment.
-
The second model represents a GLMM that accounts for site-level effects. By introducing a random intercept, factors that describe the within-site distribution of trees can be identified, independent of the absolute levels of environmental descriptors. Different preferences among taxa are analyzed within sites, and not across all sites.
-
Model 1b: Q value ~ environment + (1|site).
-
Question 2: Do site-level environmental variables predict the taxonomic composition of the populations? To answer this question, we used a GLM with mean environmental values as explanatory variables and mean Q values as response variable:
-
Model 2: mean(Q value) ~ mean(environment).
-
Question 3: Does environmental variation within sites influence the taxonomic variation of the populations? To answer this question, we used a GLM that tests the effect of variation (expressed as standard deviation, sd) in environmental predictors on the variation in Q values:
-
Model 3: sd(Q value) ~ sd(environment).
-
Question 4: Does environmental variation within sites influence the admixture levels within the populations? To answer this question, we used a GLM that tests the effect of variation (expressed as standard deviation, sd) in environmental predictors on average admixture level S of trees in a population. Model 4 is different from Model 3 because it looks at individual admixture, not population-level variation in assignment probabilities.
-
Model 4: mean(S) ~ sd(environment).
Results
Genotyping and taxonomic assignment
Of the 166 trees newly genotyped in this study, 15 were removed due to an excessive proportion of missing data. The final dataset consisted of 238 individual genotypes from 15 populations (Fig. 1). Mean Q values of Q. pubescens of the populations were between 0.36 and 0.91 (median = 0.76). Based on a Q value threshold of 0.9 as in Reutimann et al. (2020), we identified 75 Q. pubescens and seven Q. petraea trees; the remaining 156 trees were identified as presumably admixed. Individual admixture values (S) ranged from 0.08 to 0.98 (median = 0.47) and population means of S (Fig. 1) ranged from 0.29 to 0.67 (median = 0.49).
Environmental data
The scaled and centered environmental variables showed high variation within and between populations (Fig. 2). Mainly the topographic variables with a resolution of 4 m revealed ample variation within populations, which was less so for geographic position, altitude, and soil pH, the latter being based on a lower spatial resolution than the topographic variables.

Overview and distribution of the scaled and centered environmental variables used in the statistical models. Individual values are colored according to the population of origin
Statistical modelling
At the individual level, the GLM applied with a stepwise procedure for model selection (Model 1a) explained a relatively small part of the variation (R2 = 0.32, Fig. 3, Table S3). However, six topographic factors had a significant effect on Q values. The most significant variable was altitude with a negative effect, meaning that trees at high altitude were genetically less Q. pubescens-, but more Q. petraea-like. Altitude was followed by pH, vector ruggedness measure, and potential diffuse solar radiation, all having a positive effect on individual Q. pubescens Q values. Horizontal curvature and eastness had a weak but significant negative impact on the proportion of Q. pubescens in individual trees. Importance and sign of the top predictors were strongly confirmed by BMA (Fig. 4, Table S4); for example, altitude and pH, in this order, were always included in the 100 top models, followed by vector ruggedness and solar radiation. The most likely model consisted of the first three predictors.

Plot of significant model term coefficients included in the selected models and their 95% confidence intervals (*P < 0.05, **P < 0.01, and ***P < 0.001). Model 3 (sd(Q) ~ sd(E)) is not shown because it contained no significant terms. See also Table S3 for more details. Q = Q values (species assignment probabilities) of Q. pubescens; E = environmental predictors; S = admixture level; sd = standard deviation; R2 = explanatory power of the model. For details on environmental factors, see Table 1

Posterior model probabilities of the best 100 models from the Bayesian model sampling and averaging. Explanatory variables that were included in models are represented on the y-axis and cumulative model probabilities on the x-axis (models are ordered from left to right, with decreasing probabilities). Yellow and purple colors indicate that a variable is included in a particular model having a positive or negative coefficient, respectively. White color means the non-inclusion of a variable. The more important a variable is to explain the dependent variable, the more it is included in a model, resulting in a high posterior inclusion probability of the respective variable. Q = Q values (species assignment probabilities) of Q. pubescens; E = environmental predictors; S = admixture level; sd = standard deviation. For details on environmental factors, see Table 1
Adding a site identifier as random effect to the individual model (GLMM, Model 1b) led to an increased R2 of 0.45 (Fig. 3, Table S3). Hence, accounting for differences between sites revealed that other factors might be important in determining the individual taxonomic composition within populations. In addition to pH (positive), vector ruggedness (positive), and horizontal curvature (negative) that also showed a significant effect in the Model 1a without random effect, trees with a high topographic position index and potential direct solar radiation seemed to exhibit high Q. pubescens Q values within populations. For the GLMM model, no respective Bayesian model is available.
At the population level (Model 2), mean taxonomic composition seemed to be mainly driven by the more general geographic/topographic variables altitude and longitude (Fig. 3, Table S3). Populations exhibited a higher proportion of Q. pubescens again at lower altitude and in more western sites. Horizontal curvature was also included in the final model but not as a significant term, presumably not having a strong effect on the response variable. Model 2 explained the most variation of all models with R2 = 0.69. Altitude was confirmed to be a main driver of mean population Q values of Q. pubescens, as the vast majority of the posterior model mass inferred by BMA remained on models that included altitude (Fig. 4), whereas all other factors were not nearly as frequent in the top 100 models (Table S4). The most likely model included altitude, longitude and horizontal curvature.
Even though the model based on variation of environmental conditions and taxonomic composition (Model 3) showed high explanatory power (R2 = 0.53), it did not include any significant model terms (Table S3). Despite explaining a high amount of variation overall, this indicates that the variation of predictor variables has no systematic influence on the variation of Q values in our study system. PIP values of 0.47 and lower (Table S3) and no strong patterns of model inclusion of model terms in the best 100 models in the BMA support the lack of significant variables in the stepwise model selection procedure.
Finally, in Model 4, testing the effect of environmental variation on mean levels of admixture showed an R2 = 0.54 (Table S3). Admixture levels were significantly explained by the variation in topographic position index, topographic wetness index, and pH (Fig. 3, Table S3), as supported by the most likely model in BMA (Fig. 4). Whereas variation in topographic wetness index and pH was estimated to have a weak negative impact on mean admixture, variation in topographic position index had a strong positive coefficient and was also the most important variable in the BMA with a PIP of 0.63 (Table S4).
Discussion
Environmental variables are commonly used to predict the habitat suitability and distribution range of species (Elith and Leathwick 2009), the latter being potentially affected by changing environmental conditions induced by, e.g., global warming. However, this type of inference commonly neglects genetic, intraspecific variation, even though it is the foundation of species’ evolutionary processes. In this context, we show here that taxonomic composition and admixture levels of single trees and entire populations of hybridizing and sympatric white oaks (Q. petraea and Q. pubescens) are non-random, but correlating with spatial and environmental (topography and soil) variation. To our knowledge, this is the first study that uses a continuous taxonomic gradient rather than distinct species borders or taxonomic groups (Khodwekar and Gailing 2017) to model the ecological requirements of different taxa, thereby accounting for admixture among species (see also Létourneau et al. 2018, who used a similar approach with within-species genetic groups). Remarkably, we found that environmental factors cannot only explain the fine-scale taxonomic composition of admixed tree populations, but also the average admixture level of trees within these populations. Our study therefore highlights the use of species-diagnostic markers to assess intra-genera and interspecific diversity, which can then be connected to local environmental conditions to support taxonomically diverse, adapted, and resilient forest stands.
Model comparisons
To strengthen our inferences, we compared the applied model selection procedure to BMA. The latter approach is especially suited to validate the importance and sign of the different predictors while accounting for model uncertainty. Indeed, the GLMs were confirmed by BMA both in terms of sign of coefficients of the predictors and by the high congruence of PIP and P values (Table S4). The selected GLMs were always in the top three most probable models as inferred with BMA with the exception of Model 3 (testing variation) that did not include any significant terms.
Predicting the taxonomic status of single trees
Predicting the taxonomic status of individual trees can be essential in forest management and conservation. For example, when searching for seed sources for stand rejuvenation or new plantations, the selection of mother trees may be of prime importance to ensure a match between the micro-environmental conditions at source and target location. Similarly, knowledge about the taxonomy of single trees might be relevant in selective tree removal in forest management to promote adapted and resilient forest stands. However, this study shows that such ambitious goals might be difficult to achieve, because the explanatory power of Model 1a (R2 = 0.32) was only moderate. The following issues could explain this outcome. First, the applied taxonomic assignment might result in some noise that cannot be explained by environmental variation. Second, our environmental predictors (including the exact location of the individual trees) might not be accurate enough to describe the microhabitat of single trees. Finally, stochastic processes and unmeasured factors like human influence (e.g., forest management), differences in phenology, animal dispersal vectors, wind patterns, and competition might substantially influence the spatial arrangement of single trees. The effect of competition is supported by the improvement with model 1b (R2 = 0.45) that includes a random factor for site (see below).
Still, there were six environmental predictors that had a significant effect on the taxonomic status of individual trees (Model 1a). Most importantly, oaks with a high proportion of Q. pubescens in their genome were found at low altitude and high soil pH. Whereas it has been shown that Q. pubescens prefers more alkaline, calcareous soils (Leroy et al. 2020; Rellstab et al. 2016b), it is not clear whether the link to low altitude comes from its tolerance for very warm conditions (Leroy et al. 2020) or whether our sampling setup partly ignored populations of Q. pubescens at high altitudes, where this species is known to occur. Of the topographic factors, topographic heterogeneity (ruggedness) and direct solar radiation had the strongest effect and were correlated to higher proportions of Q. pubescens in single trees, the latter confirming a previous result in a large-scale study at the population level (Rellstab et al. 2016b) and likely being related to soil temperature and water regime. When we accounted for site effects by introducing a random factor (Model 1b), the strong and significant effect of altitude disappeared, showing that the effect of altitude was mainly driven at the site level. Those predictors that remain significant and strong (in this order: pH, ruggedness, diffuse solar radiation) best explain taxonomic variation within sites and could be seen as the factors that are important in competition of the two taxa in occupying different micro-environments within the sites. Going further, the individual-based models could be improved by in situ measurements of soil and atmospheric conditions, which are known to be highly fluctuating locally on a daily basis in mountainous regions.
Predicting the taxonomic composition of populations
Since stochastic and unmeasured effects limit our ability to predict the taxonomic status of single trees, population-level analyses might be more promising in describing the ecological preferences of the studied taxa along the continuous taxonomic gradient. Indeed, population-level analyses of taxonomic composition and environmental descriptors had a very high explanatory power (Model 2, R2 = 0.69). Averaging both components obviously removed possible noise associated to the uncertainties discussed in the single-tree models. Again, altitude was the strongest descriptor in the final model, with higher genetic fractions of Q. pubescens occurring in populations at lower mean altitude. Moreover, longitude was a significant descriptor, with lower proportions of Q. pubescens in more eastern populations. Such an effect of geographical location could come from correlation with other environmental descriptors (specifically topographic position index in this case) or unmeasured variables (e.g., predominant wind direction). Overall, the population-level analyses partly confirm the results of the individual models, but with less significant terms.
Predicting the taxonomic variation of populations
Because different oak species have different ecological requirements and tolerances, taxonomically diverse oak stands should be well equipped for adaptation to environmental changes through natural selection and adaptive introgression (Lazic et al. 2021) and be more resilient to extreme events. Here, we hypothesized that variation in micro-environmental conditions promotes such high taxonomic diversity, and that certain environmental factors are driving this pattern. However, despite high explanatory power (R2 = 0.53), we did not find any significant predictors in Model 3. In other words, no predictor had a consistent influence on taxonomic diversity. This points to complex processes that determine diversity in these sites such as community dynamics, species interactions, and colonization history.
Predicting the admixture levels within populations
Finally, we tested whether variation in micro-environmental conditions can predict the mean admixture level of individuals within a population (Model 4). This model is different to Model 3, in which taxonomic diversity is defined as variation within populations, and not within individuals. All populations considered in this study showed relatively high levels of admixture (Fig. 1), and the explanatory power of Model 4 was high (R2 = 0.54). Variation in topographic position index (the difference between the elevation of the focal tree and the mean of its surrounding environment) was substantially and positively correlated with admixture levels. Topographic position index is a proxy for topographic exposure (the difference of the tree’s elevation to the mean elevation of a specified neighborhood), and high values might be linked to extreme temperatures and low soil water content. Habitat heterogeneity could be linked to admixture levels in two ways. First, heterogeneous habitats might represent a fitness advantage for admixed individuals, because they are genetically more diverse and therefore might be better equipped for a variety of environmental conditions than their more specialized parental species. Alternatively, elevational heterogeneity might promote admixture processes in mixed oak stands by enhancing pollen flow within and between sites, similarly as fire-induced population thinning increased gene flow in Pinus halepensis (Shohami and Nathan 2014). Contrary, variation in pH and topographic wetness index actually seem to impede admixture processes, shown by weak negative correlation with admixture index S. However, the exact processes from which such negative correlations can arise remain unknown.
Implications
In a world of rapid climate change, forest tree species are especially challenged. Due to their long lifespan and generation time (Petit and Hampe 2006), they can only slowly adapt to new conditions (Aitken et al. 2008; Dauphin et al. 2021) and often show limited dispersal abilities (heavy seeds, animal vectors) to track their present ecological niche. However, they normally exhibit very high genetic diversity due to their large population sizes and massive gene flow by wind pollination (Petit and Hampe 2006). In large populations, natural selection is highly efficient, and adaptational shifts can be achieved in one generation (Saleh et al. 2022). Gene flow allows the introgression of gene variants that could be beneficial in future or changing environments, but can also counteract genetic adaptation (Kremer et al. 2012). In this sense, hybridization and admixture between species, as studied here, can be an important component for forests and their present species. We show here that on local and regional scale, a large, continuous taxonomic gradient within Quercus sect. Quercus exists. Both species can be found as pure individuals, but the populations are clearly dominated by admixed trees. The spatial arrangement of these continuous taxa is not random, but partly correlated to (micro-) environmental conditions. Similar findings of a non-random association of taxonomic gradients and environmental conditions have been shown in two sympatric red oak species (Khodwekar and Gailing 2017). This clearly shows that white oak species should not be considered and managed as pure species, but be seen as a highly diverse species complex with a high adaptive potential through hybridization and introgression. Our data also suggest that habitat heterogeneity promotes admixture processes that normally result in increased genetic diversity—an important aspect to ensure resilient and adaptive forests for ongoing and forthcoming environmental changes. Finally, from a scientific perspective, incorporating information about evolutionary lineages, subspecies, or admixture into SDMs can substantially increase the precision of a species’ predicted tolerance to climate change (Lecocq et al. 2019; Razgour et al. 2019; Waldvogel et al. 2020).
Limitations
Our study has important limitations that need to be considered when interpreting the results. First of all, our inferences are purely based on correlative approaches that do not necessarily describe causality. This is especially true for the topographic variables used in the model that are also proxies for micro-climatic conditions. Moreover, these topographic characteristics mostly stay constant over time and do not allow for future projections, unless they are somehow linked to terrain movements or climatic data. However, climate projections do not have the required resolution to work on such a scale. The only alternative are in situ measurements, which should be considered in future studies looking at micro-environmental variation. Second, the findings are limited to our regional sampling area; inferences on which environmental factors are generally driving the two species and their admixture should be treated with caution. Within our study region, the selected sites only partly represent the environmental ranges of the two species. Still, many of our findings, for example, regarding temperature and pH, are actually congruent with previous findings on a larger scale (Leroy et al. 2020; Rellstab et al. 2016b). Finally, it is important to again emphasize that the species-diagnostic SNPs used here represent taxonomic, and not necessarily neutral genetic diversity (Reutimann et al. 2020). Although it is very likely that levels of admixture are highly linked to genetic diversity, other markers—preferably genome-wide and much higher in numbers (Fischer et al. 2017)—are needed to accurately describe genetic diversity, especially within taxa.
Conclusions
Our results show that a multitude of environmental factors shapes the taxonomic and admixture landscape of these two hybridizing oak species on local and regional scale and that heterogeneity of these factors promote taxonomic diversity and admixture. These outcomes also highlight the need for individual, in situ measured variables to even better explain the potential drivers of fine-scale patterns of individual taxonomic status and genetic composition. Overall, our study stresses the prospects of studying high-resolution environmental factors and detailed taxonomic gradients with genetic data for understanding the ecological and evolutionary processes that shape the local taxonomic compositions and levels of admixture. Information on taxonomic gradients can greatly complement SDMs, which are mostly based on presences and absences of uniform species and therefore often ignore the existence of admixed individuals, several ecotypes, and their ecological niches.
References
Abbott RJ (2017) Plant speciation across environmental gradients and the occurrence and nature of hybrid zones. J Syst Evol 55:238–258. https://doi.org/10.1111/jse.12267
Aitken SN, Yeaman S, Holliday JA, Wang TL, Curtis-McLane S (2008) Adaptation, migration or extirpation: climate change outcomes for tree populations. Evol Appl 1:95–111. https://doi.org/10.1111/j.1752-4571.2007.00013.x
Baltensweiler A, Walthert L, Hanewinkel M, Zimmermann S, Nussbaum M (2021) Machine learning based soil maps for a wide range of soil properties for the forested area of Switzerland. Geoderma Reg 27 https://doi.org/10.1016/j.geodrs.2021.e00437
Bruschi P, Vendramin GG, Bussotti F, Grossoni P (2000) Morphological and molecular differentiation between Quercus petraea (Matt.) Liebl. and Quercus pubescens Willd. (Fagaceae) in northern and central Italy. Ann Bot 85:325–333. https://doi.org/10.1006/anbo.1999.1046
Cochard H, Breda N, Granier A, Aussenac G (1992) Vulnerability to air-embolism of 3 European oak species (Quercus petraea (Matt) Liebl, Q pubescens Willd, Q robur L). Ann Sci Forest 49:225–233. https://doi.org/10.1051/orest:19920302
Conrad O et al (2015) System for automated geoscientific analyses (SAGA) v. 2.1.4. Geosci Model Dev 8:1991–2007. https://doi.org/10.5194/gmd-8-1991-2015
Cruzan MB, Arnold ML (1993) Ecological and genetic associations in an Iris hybrid zone. Evolution 47:1432–1445. https://doi.org/10.2307/2410158
Curtu AL, Gailing O, Finkeldey R (2007) Evidence for hybridization and introgression within a species-rich oak (Quercus spp.) community. BMC Evol Biol 7:218. https://doi.org/10.1186/1471-2148-7-218
Curtu AL, Gailing O, Leinemann L, Finkeldey R (2007) Genetic variation and differentiation within a natural community of five oak species (Quercus spp.). Plant Biol 9:116–126. https://doi.org/10.1055/s-2006-924542
Curtu AL, Gailing O, Finkeldey R (2009) Patterns of contemporary hybridization inferred from paternity analysis in a four-oak-species forest. BMC Evol Biol 9 https://doi.org/10.1186/1471-2148-9-284
Dauphin B et al (2021) Genomic vulnerability to rapid climate warming in a tree species with a long generation time. Glob Change Biol 27:1181–1195. https://doi.org/10.1111/gcb.15469
Douma JC, Weedon JT (2019) Analysing continuous proportions in ecology and evolution: a practical introduction to beta and Dirichlet regression. Methods Ecol Evol 10:1412–1430. https://doi.org/10.1111/2041-210x.13234
Elith J, Leathwick JR (2009) Species distribution models: Ecological explanation and prediction across space and time. Annu Rev Ecol Evol S 40:677–697. https://doi.org/10.1146/annurev.ecolsys.110308.120159
Fischer MC et al (2017) Estimating genomic diversity and population differentiation – an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genomics 18:69. https://doi.org/10.1186/s12864-016-3459-7
Fox J, Weisberg HS (2019) An R companion to applied regression. Sage Publications
Fu RR et al (2022) Genome-wide analyses of introgression between two sympatric Asian oak species. Nat Ecol Evol 6:924–935. https://doi.org/10.1038/s41559-022-01754-7
Galle A, Haldimann P, Feller U (2007) Photosynthetic performance and water relations in young pubescent oak (Quercus pubescens) trees during drought stress and recovery. New Phytol 174:799–810. https://doi.org/10.1111/j.1469-8137.2007.02047.x
Gerber S, Chadoeuf J, Gugerli F, Lascoux M, Buiteveld J, Cottrell J, Dounavi A, Fineschi S, Forrest LL, Fogelqvist J, Goicoechea PG, Jensen JS, Salvini D, Vendramin GG, Kremer A (2014) High rates of gene flow by pollen and seed in oak populations across Europe. PLOS ONE 9:e91301. https://doi.org/10.1371/journal.pone.0091301
Gugerli F, Brodbeck S, Holderegger R (2008) Utility of multilocus genotypes for taxon assignment in stands of closely related European white oaks from Switzerland. Ann Bot 102:855–863. https://doi.org/10.1093/aob/mcn164
Guichoux E, Lagache L, Wagner S, Léger P, Petit RJ (2011) Two highly validated multiplexes (12-plex and 8-plex) for species delimitation and parentage analysis in oaks (Quercus spp.). Mol Ecol Resour 11:578–585. https://doi.org/10.1111/j.1755-0998.2011.02983.x
Guisan A, Thuiller W, Zimmermann NE (2017) Habitat suitability and distribution models: With applications in R. Cambridge University Press, Ecology, Biodiversity and Conservation
Hamilton JA, De la Torre AR, Aitken SN (2015) Fine-scale environmental variation contributes to introgression in a three-species spruce hybrid complex. Tree Genet Genomes 11:817. https://doi.org/10.1007/s11295-014-0817-y
Hijmans RJ (2019) Raster: Geographic data analysis and modeling. R package. https://cran.r-project.org/web/packages/raster/index.html
Johnston JA, Wesselingh RA, Bouck AC, Donovan LA, Arnold ML (2001) Intimately linked or hardly speaking? The relationship between genotype and environmental gradients in a Louisiana Iris hybrid population. Mol Ecol 10:673–681. https://doi.org/10.1046/j.1365-294x.2001.01217.x
Kampfer S, Lexer C, Glossl J, Steinkellner H (1998) Characterization of (GA)(n) microsatellite loci from Quercus robur. Hereditas 129:183–186. https://doi.org/10.1111/j.1601-5223.1998.00183.x
Khodwekar S, Gailing O (2017) Evidence for environment-dependent introgression of adaptive genes between two red oak species with different drought adaptations. Am J Bot 104:1088–1098. https://doi.org/10.3732/ajb.1700060
Körner C, Riedl S (2021) Alpine treelines: functional ecology of the global high elevation tree limits vol, 3rd edn. Springer
Kremer A et al (2012) Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecol Lett 15:378–392. https://doi.org/10.1111/j.1461-0248.2012.01746.x
Kuhn M (2008) Building predictive models in R using the caret package. J Stat Softw 28:1–26. https://doi.org/10.18637/jss.v028.i05
Lazic D, Hipp AL, Carlson JE, Gailing O (2021) Use of genomic resources to assess adaptive divergence and introgression in oaks. Forests 12:690. https://doi.org/10.3390/f12060690
Lecocq T, Harpke A, Rasmont P, Schweiger O (2019) Integrating intraspecific differentiation in species distribution models: Consequences on projections of current and future climatically suitable areas of species. Divers Distrib 25:1088–1100. https://doi.org/10.1111/ddi.12916
Leempoel K, Parisod C, Geiser C, Daprà L, Vittoz P, Joost S (2015) Very high resolution digital elevation models: are multi-scale derived variables ecologically relevant? Methods Ecol Evol 6:1373–1383. https://doi.org/10.1111/2041-210X.12427
Lefèvre F, Pichot C, Beuker E, Kowalczyk J, Matras J, Ziehe M, Villar M, Peter M, Gugerli F, Orazio C, Cordero Montoya R, Alia R, Van Halder I, González-Martínez SC, Kremer A (2016) Intensive study sites. In: Evolution of trees and forest communities. PG Edition, Bordeaux, pp 6–9
Lepais O, Gerber S (2011) Reproductive patterns shape introgression dynamics and species succession within the European white oak species complex. Evolution 65:156–170. https://doi.org/10.1111/j.1558-5646.2010.01101.x
Leroy T et al (2017) Extensive recent secondary contacts between four European white oak species. New Phytol 214:865–878. https://doi.org/10.1111/nph.14413
Leroy T et al (2020) Massive postglacial gene flow between European white oaks uncovered genes underlying species barriers. New Phytol 226:1183–1197. https://doi.org/10.1111/nph.16039
Létourneau J, Ferchaud AL, Le Luyer J, Laporte M, Garant D, Bernatchez L (2018) Predicting the genetic impact of stocking in Brook Charr (Salvelinus fontinalis) by combining RAD sequencing and modeling of explanatory variables. Evol Appl 11:577–592. https://doi.org/10.1111/eva.12566
Neophytou C (2014) Bayesian clustering analyses for genetic assignment and study of hybridization in oaks: effects of asymmetric phylogenies and asymmetric sampling schemes. Tree Genet Genomes 10:273–285. https://doi.org/10.1007/s11295-013-0680-2
Petit RJ, Hampe A (2006) Some evolutionary consequences of being a tree. Annu Rev Ecol Evol S 37:187–214. https://doi.org/10.1146/annurev.ecolsys.37.091305.110215
Petit RJ, Kremer A, Wagner DB (1993) Geographic structure of chloroplast DNA polymorphisms in European oaks. Theor Appl Genet 87:122–128. https://doi.org/10.1007/Bf00223755
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Development Core Team (2020) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Randler C (2007) Assortative mating of Carrion Corvus corone and Hooded Crows C. cornix in the hybrid zone in eastern Germany. Ardea 95:143–149. https://doi.org/10.5253/078.095.0116
Razgour O et al (2019) Considering adaptive genetic variation in climate change vulnerability assessment reduces species range loss projections. P Natl Acad Sci USA 116:10418–10423. https://doi.org/10.1073/pnas.1820663116
Rellstab C, Bühler A, Graf R, Folly C, Gugerli F (2016) Using joint multivariate analyses of leaf morphology and molecular-genetic markers for taxon identification in three hybridizing European white oak (Quercus spp.) species. Ann for Sci 73:669–679. https://doi.org/10.1007/s13595-016-0552-7
Rellstab C et al (2016) Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol Ecol 25:5907–5924. https://doi.org/10.1111/mec.13889
Reutimann O, Gugerli F, Rellstab C (2020) A species-discriminatory single-nucleotide polymorphism set reveals maintenance of species integrity in hybridizing European white oaks (Quercus spp.) despite high levels of admixture. Ann Bot 125:663–676. https://doi.org/10.1093/aob/mcaa001
Rigby RA, Stasinopoulos DM (2005) Generalized additive models for location, scale and shape. J R Stat Soc C-Appl 54:507–544. https://doi.org/10.1111/j.1467-9876.2005.00510.x
Saleh D et al (2022) Genome-wide evolutionary response of European oaks during the Anthropocene. Evol Lett 6:4–20. https://doi.org/10.1002/evl3.269
Salvini D, Bruschi P, Fineschi S, Grossoni P, Kjaer ED, Vendramin GG (2009) Natural hybridisation between Quercus petraea (Matt.) Liebl. and Quercus pubescens Willd. within an Italian stand as revealed by microsatellite fingerprinting. Plant Biol 11:758–765. https://doi.org/10.1111/j.1438-8677.2008.00158.x
Shohami D, Nathan R (2014) Fire-induced population reduction and landscape opening increases gene flow via pollen dispersal in Pinus halepensis. Mol Ecol 23:70–81. https://doi.org/10.1111/mec.12506
Steinkellner H et al (1997) Identification and characterization of (GA/CT)(n)-microsatellite loci from Quercus petraea. Plant Mol Biol 33:1093–1096. https://doi.org/10.1023/A:1005736722794
Stierschneider M, Gaubitzer S, Schmidt J, Weichselbaum O, Kopecky D, Kremer A, Fluch S, Sehr EM (2016) The Evoltree Repository Centre – A central access point for reference material and data of forest genetic resources. In: Kremer A, Hayes S, González-Martínez SC (eds) Evolution of Trees and forest communities: ten years of the evoltree network. PG Edition, Bordeaux, pp 15–19
Suarez-Gonzalez A, Lexer C, Cronk QCB (2018) Adaptive introgression: a plant perspective. Biol Letters 14:20170688. https://doi.org/10.1098/rsbl.2017.0688
Truffaut L, Chancerel E, Ducousso A, Dupouey JL, Badeau V, Ehrenmann F, Kremer A (2017) Fine-scale species distribution changes in a mixed oak stand over two successive generations. New Phytol 215:126–139. https://doi.org/10.1111/nph.14561
Vijay N, Bossu CM, Poelstra JW, Weissensteiner MH, Suh A, Kryukov AP, Wolf JBW (2016) Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat Commun 7:13195. https://doi.org/10.1038/ncomms13195
Viscosi V, Lepais O, Gerber S, Fortini P (2009) Leaf morphological analyses in four European oak species (Quercus) and their hybrids: a comparison of traditional and geometric morphometric methods. Plant Biosyst 143:564–574. https://doi.org/10.1080/11263500902723129
Waldvogel AM et al (2020) Evolutionary genomics can improve prediction of species’ responses to climate change. Evol Lett 4:4–18. https://doi.org/10.1002/evl3.154
Zeugner S (2011) Bayesian model averaging with BMS. Tutorial to the R-package BMS 1e30
Zeugner S, Feldkircher M (2015) Bayesian model averaging employing fixed and flexible priors: the BMS package for R. J Stat Softw 68:1–37
Zuur AF, Ieno EN, Elphick CS (2010) A protocol for data exploration to avoid common statistical problems. Methods Ecol Evol 1:3–14. https://doi.org/10.1111/j.2041-210X.2009.00001.x
Acknowledgements
We thank LGC Genomics for SNP genotyping, AIT Vienna for supplying DNA samples from the EvolTree repository, René Graf for lab work, EvolTree for financing the sampling campaign in the course of establishing permanent genetic resources for EvolTree ISSs, everybody who helped in the field when sampling leaves, and two anonymous reviewers for their valuable input. This study is part of the project OakID2 and was financed by the EvolTree Opportunity call.
Funding
Open Access funding provided by Lib4RI – Library for the Research Institutes within the ETH Domain: Eawag, Empa, PSI & WSL.
Author information
Authors and Affiliations
Contributions
CR and AK acquired funding; OR and CR developed the concept; OR, BD, and AB analyzed the data; OR and CR wrote the manuscript, with substantial input from all co-authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Data archiving statement
Raw geographic and environmental data, and genotypic information are available as supplementary material. The R-code used for the analysis is available on request.
Additional information
Communicated by J. Chen
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reutimann, O., Dauphin, B., Baltensweiler, A. et al. Abiotic factors predict taxonomic composition and genetic admixture in populations of hybridizing white oak species (Quercus sect. Quercus) on regional scale. Tree Genetics & Genomes 19, 22 (2023). https://doi.org/10.1007/s11295-023-01598-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-023-01598-7