OGG1 Inhibitor TH5487 Alters OGG1 Chromatin Dynamics and Prevents Incisions


1
Science for Life Laboratory, Department of Oncology-Pathology, Karolinska Institutet, 171 77 Stockholm, Sweden
2
Weston Park Cancer Centre, Department of Oncology and Metabolism, University of Sheffield, Sheffield S10 2RX, UK
*
Author to whom correspondence should be addressed.
Biomolecules 2020, 10(11), 1483; https://doi.org/10.3390/biom10111483
Submission received: 14 September 2020
/
Revised: 11 October 2020
/
Accepted: 21 October 2020
/
Published: 26 October 2020
(This article belongs to the Section Molecular Biology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:8-oxoguanine DNA glycosylase (OGG1) is the main DNA glycosylase responsible for the excision of 7,8-dihydro-8-oxoguanine (8-oxoG) from duplex DNA to initiate base excision repair. This glycosylase activity is relevant in many pathological conditions including cancer, inflammation, and neurodegenerative diseases. To have a better understanding of the role of OGG1, we previously reported TH5487, a potent active site inhibitor of OGG1. Here, we further investigate the consequences of inhibiting OGG1 with TH5487. TH5487 treatment induces accumulation of genomic 8-oxoG lesions. Furthermore, it impairs the chromatin binding of OGG1 and results in lower recruitment of OGG1 to regions of DNA damage. Inhibiting OGG1 with TH5487 interferes with OGG1′s incision activity, resulting in fewer DNA double-strand breaks in cells exposed to oxidative stress. This study validates TH5487 as a potent OGG1 inhibitor that prevents the repair of 8-oxoG and alters OGG1–chromatin dynamics and OGG1′s recruitment kinetics.
1. Introduction
Reactive oxygen species (ROS) can be generated from a number of endogenous or exogenous sources. Endogenously generated ROS are a by-product of normal oxygen metabolism in different organelles including mitochondria, peroxisomes, and the endoplasmic reticulum. Exposure to ionizing radiation or some chemicals are among the exogenous sources of ROS [1]. Multiple lines of evidence indicate that loss of balanced redox homeostasis is implicated in many pathological conditions, e.g. cancer, neurodegenerative diseases, cardiovascular diseases, ischemia reperfusion injury, and inflammatory diseases, in addition to aging [2,3,4,5,6]. When maintained at appropriate cellular concentrations, ROS are involved in several physiological processes such as signaling pathways and immune responses [3,7]. However, at higher concentrations, ROS can overwhelm the cellular antioxidant capacity, generating oxidative stress and causing oxidative damage to lipids, proteins and DNA [1,3,4].
Owing to its low redox potential, guanine is the most vulnerable nucleobase to oxidation [8,9,10]. The major product of guanine oxidation is 7,8-dihydro-8-oxoguanine (8-oxoG), a mutagenic lesion that can mispair to adenine, leading to base transversions and potential genetic mutations [11]. 8-oxoguanine DNA glycosylase (OGG1) is the main glycosylase catalyzing 8-oxoG excision from duplex DNA when it is paired to cytosine to initiate base excision repair (BER) [12,13]. Recently, we reported the development of TH5487, a potent small-molecule active-site OGG1 inhibitor and provided proof of concept that targeting oxidative DNA repair by inhibiting OGG1 might serve as a beneficial strategy to alleviate inflammation [14].
Here, we aimed to further characterize TH5487 in terms of its capacity to inhibit DNA repair. In addition, we evaluated the inhibitor’s effect on OGG1–chromatin dynamics and how it impacts DNA incision. We observed that TH5487 treatment results in the accumulation of genomic 8-oxoG lesions. Furthermore, TH5487 reduces OGG1–chromatin binding and results in lower recruitment of OGG1 to regions of DNA damage. Inhibiting OGG1 with TH5487 impairs OGG1′s incision activity, resulting in fewer DNA double-strand breaks in oxidatively stressed cells. Taken together, our results indicate that TH5487 inhibits OGG1, preventing the repair of 8-oxoG, and alters OGG1–chromatin dynamics and OGG1′s recruitment kinetics.
2. Materials and Methods
2.1. Cell Culture and Treatments
U2OS cells were cultured at 37 °C in a 5% CO2 atmosphere in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 10% of fetal bovine serum (FBS; Gibco) and 100 U/mL penicillin–streptomycin (Gibco). To induce oxidative DNA damage, cells at about 80% confluency were treated with potassium bromate (Sigma-Aldrich CAS 7758-01-2) in serum-free DMEM for the indicated period of time. To inhibit OGG1, 10 μM TH5487 or 0.1% dimethyl sulfoxide (DMSO; VWR Chemicals) was used according to the indicated experimental scheme for the appropriate time periods.
2.2. Generation of Stable Cell Lines Expressing OGG1-GFP
U2OS cells were transfected with the hOGG1-GFP vector described in Visnes et al. [14] using jetPEI (Polyplus) and selected with 1 μg/mL puromycin for 10 days. To minimize variability in the expression levels, clonal expansion was carried out to generate a single clone of U2OS cells constitutively expressing GFP-tagged OGG1.
2.3. 8-oxoG Immunofluorescence Assay
Cells were seeded on an Ibidi 6-channel µ-Slide VI 0.4 (Ibidi, Catalog No. 80606) 24 h prior to the indicated treatment. Next, 20 mM potassium bromate (KBrO3; Sigma-Aldrich CAS 7758-01-2) was prepared in serum-free medium and added to the cells for 1 hour. Cells were then allowed to recover in serum-free medium containing either 0.1% DMSO or 10 μM TH5487 for 1, 4, or 6 hours. Cells were fixed with ice-cold acetone:methanol 1:1 (Fisher Scientific) for 20 min on ice and permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 5 min. This was followed by RNAse treatment to degrade RNAs using the following buffer: 10 mM Tris-HCl (pH 7.5), 1 mM Ethylenediaminetetraacetic acid (EDTA), 0.4 mM NaCl, and 100 μg/mL RNAse (Zymo Research) for 1 hour at 37 °C. To make the 8-oxoG lesions accessible to the antibody, the DNA duplex was denatured by incubating the cells in freshly prepared 2.5 N HCl for 30 min at room temperature followed by a neutralization step using 0.1 M sodium borate Na2Bo4O7 (pH 8.8) for 10 min. Blocking with 4% bovine serum albumin (BSA; Sigma) in PBS was carried out for 1 h, followed by overnight incubation with anti-8-OHdG (Abcam, AB48508, N45.1) at 1:200. Incubation with the secondary antibody Alexa 647 (Thermo Fisher Scientific) was performed for 1 h at room temperature. 4′,6-diamidino-2-phenylindole (DAPI) was used to stain DNA. Imaging was performed with a Zeiss LSM 780 confocal microscope equipped with a UV-transmitting Plan-Apochromat 40×/1.30 Oil DIC M27 objective. The “tile scanning” and “positions” tools of ZEN software (ZEN, Zeiss, Germany) were used to acquire images from at least 9 positions per condition (unidirectional scanning, zoom 1, scan speed 10). Images were processed in ImageJ and Cell Profiler. For quantitative evaluation of 8-oxoG levels, the data of at least 500 cells from three independent experiments were averaged. The mean fluorescence intensity and the standard deviation were calculated and displayed using GraphPad Prism software.
2.4. In Situ Extraction
To study OGG1 retention under different experimental conditions, a pre-extraction treatment was performed to wash off soluble proteins loosely bound to the chromatin. U2OS cells stably expressing GFP-tagged OGG1 were treated with 0.1% Triton X-100 in PBS (Sigma) for 1 min prior to fixation with 4% paraformaldehyde (Santa Cruz). DAPI was used to stain the DNA for 10 min. Cells were then examined under a Zeiss LSM 780 confocal microscope equipped with a UV-transmitting Plan-Apochromat 63x/1.40 Oil DIC M27 objective. A 488 nm Ar laser was used to excite GFP. OGG1-GFP nuclear fluorescence signal intensities were recorded from three independent experiments. Images were processed in ImageJ and Cell Profiler. Data from at least 1300 cells for each treatment condition were assessed. The mean fluorescence intensity and the standard deviation were calculated and displayed using GraphPad Prism software.
2.5. Live Cell Microscopy, Laser Microirradiation and Fluorescence Recovery after Photobleaching
Live cell microscopy, microirradiation and fluorescence recovery after photobleaching (FRAP) experiments were performed as previously described by Xie et al. [15] and Visnes et al. [14]. In brief, U2OS cells stably expressing GFP-tagged wild-type OGG1 were seeded on Ibidi µ-dish (Ibidi #81166) 24 h prior to the indicated treatment. For laser microirradiation, cells were pre-sensitized with 10 μg/mL Hoechst 33342 (Thermo Fisher Scientific, Catalog No. 62249) for 10 min at 37 °C. To avoid background fluorescence from phenol red present in the DMEM culture medium, we exchanged the medium to live cell imaging medium (Thermo Fisher Scientific, Catalog No. 31053028) supplemented with penicillin–streptomycin antibiotics, 10% FBS and 25 mM HEPES containing either 0.1% DMSO or 10 μM TH5487 for 1 h. Cells were then transferred to a 37 °C pre-heated environmental chamber attached to a Zeiss LSM 780 confocal microscope equipped with a UV-transmitting Plan-Apochromat 40×/1.30 Oil DIC M27 objective. To induce DNA damage, a nuclear spot (dimensions: 10 × 10 pixels) was selected using the circular region tool of the ZEN software (ZEN, Zeiss, Germany) and irradiated using a 405 nm diode laser set to 100% (spot irradiation, 1 iteration, zoom 5, and pixel dwell time of 12.61 µs). For quantitative evaluation of the recruitment kinetics, the fluorescence intensity at the irradiated spot was corrected for background and for total nuclear loss of fluorescence over the time course and normalized to the pre-irradiation value.
To induce oxidative stress for FRAP experiments, cells were challenged for 1 h with 40 mM potassium bromate (Sigma-Aldrich CAS 7758-01-2) dissolved in live cell imaging medium supplemented with penicillin–streptomycin antibiotics, 10% FBS and 25 mM HEPES (Gibco) as a pH buffer. This was followed by adding 0.1% DMSO or 10 μM TH5487 to the medium for another hour. The FRAP assay was performed according to the protocol described by Visnes et al. [14]. The data of at least 35 nuclei from three independent experiments were averaged. Images were processed in ImageJ. The mean curve and the standard error of the mean were calculated and displayed using GraphPad Prism software.
2.6. Quantitative Microscopy
For quantitative microscopy, cells were seeded in 96-well plates (10,000 cells/well; Corning 4680). Cells were treated with either 40 mM potassium bromate or 50 μM menadione and increasing concentrations of TH5487 or equal amounts of DMSO in serum-free DMEM. After 1 h of incubation, cells were pre-extracted with 0.1% Tritonx-100 in PBS for 1 min, fixed in 4% formaldehyde for 20 min, permeabilized with 0.5% Triton X-100 for 10 min, and probed with an anti-γH2AX antibody (Millipore, 05-636). Images were taken with an Image Xpress Micro (Molecular Devices) microscope using a 20× lens. Fluorescence intensities per cell nucleus were determined using a pipeline generated in Cell Profiler software and plotted using GraphPad Prism.
2.7. Statistical Analysis
Data from two to three independent experiments were subjected to a two-tailed Student’s t-test to determine the statistical significance and are presented as means ± standard error of the mean (SEM) or standard deviation (SD) as indicated.
3. Results
3.1. TH5487 Treatment Results in Accumulation of Genomic 8-OxoG Lesions
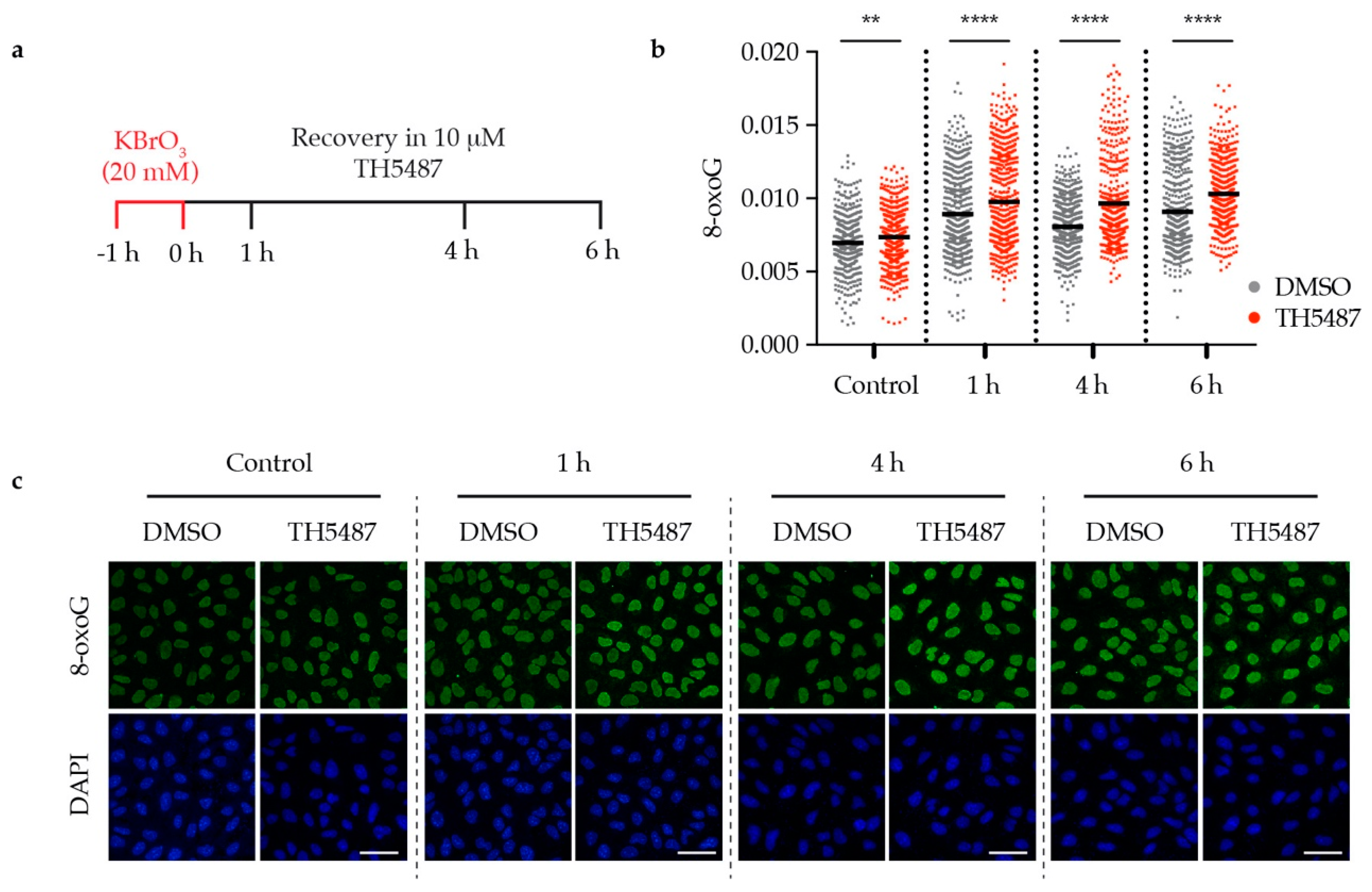
We have previously described TH5487 as a selective small molecule inhibitor of OGG1 [14]. To further study the repair kinetics of 8-oxoG after inhibiting OGG1, we used the oxidizing agent potassium bromate (KBrO3), which has been reported to induce DNA oxidative damage, resulting in 8-oxoG lesions [16,17]. U2OS (osteosarcoma) cancer cells were pre-treated with 20 mM KBrO3 for 1 hour then released into fresh medium containing either 10 μM TH5487 or 0.1% DMSO. The levels of genomic 8-oxoG were found to be significantly higher in TH5487-treated cells at all time points, indicating that TH5487 prevents OGG1 from repairing its main substrate, 8-oxoG (Figure 1a–c).
3.2. TH5487 Treatment Impairs OGG1 Binding to Damaged Chromatin
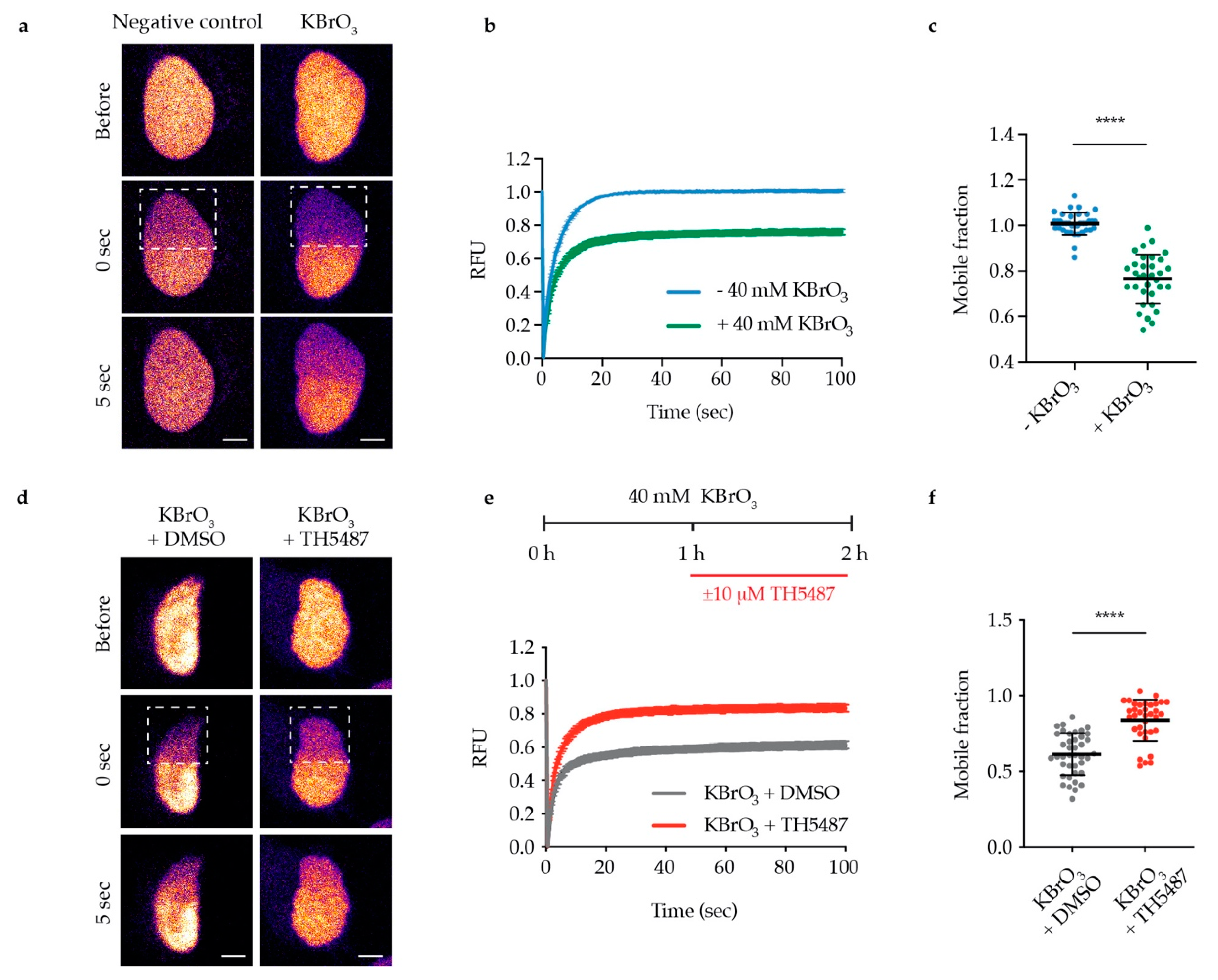
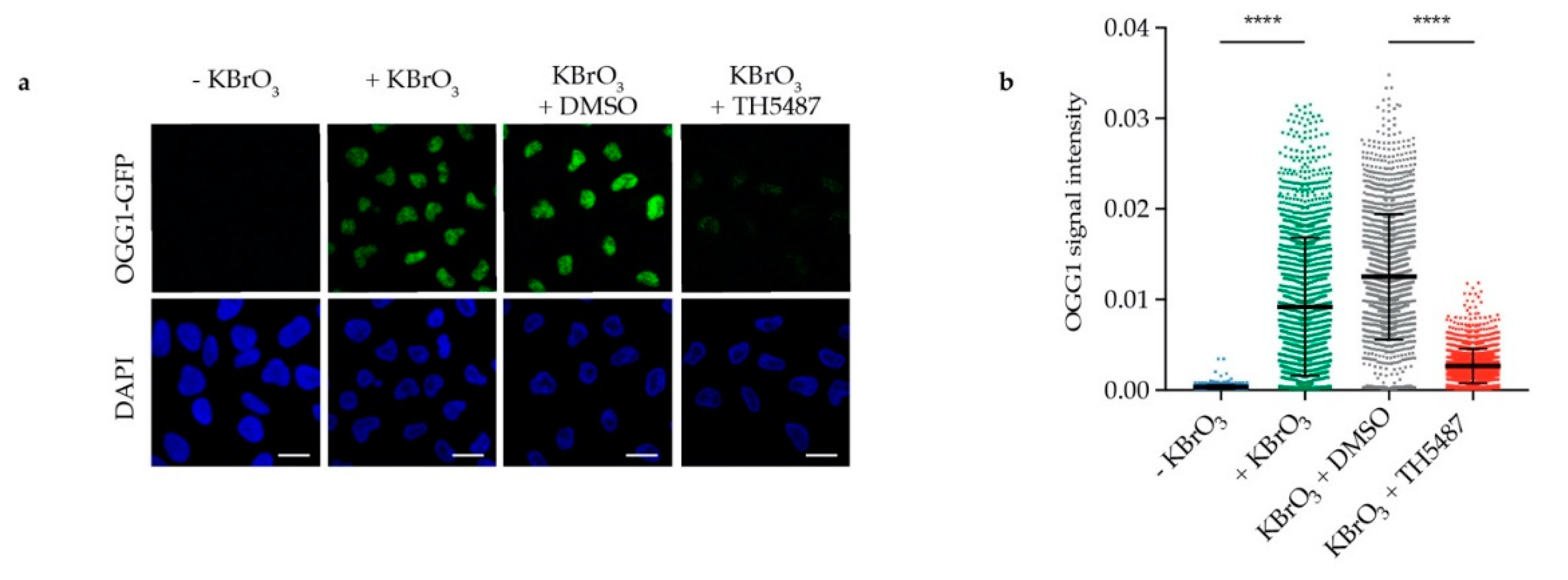
The observed reduction in OGG1′s repair activity might arise from OGG1 being trapped on DNA. Alternatively, it might be due to OGG1 being unable to bind 8-oxoG because of its active site being occupied by TH5487. To test these hypotheses, we examined OGG1–chromatin binding using fluorescence recovery after photobleaching (FRAP) in U2OS cells stably expressing GFP-tagged OGG1. We observed strong OGG1 binding to chromatin after KBrO3 treatment (Figure 2a–c), indicating that such treatment results in oxidative DNA damage, to which OGG1 binds. Interestingly, OGG1 becomes more mobile in cells co-treated with KBrO3 and TH5487 (Figure 2d–f), suggesting that TH5487 prevents OGG1 from binding to damaged DNA. Potassium bromate has been shown to induce OGG1 retention on chromatin after in situ extraction [18]. Consistent with the FRAP results, more OGG1-GFP was found to be retained in U2OS cells treated with KBrO3 following in situ extraction, though this was reversed after concomitant treatment with KBrO3 and TH5487 (Figure 3). Taken together, these results show that TH5487 treatment impairs OGG1 binding to damaged chromatin.
3.3. OGG1 Recruitment to Laser-Induced DNA Damage Sites is Reduced by TH5487
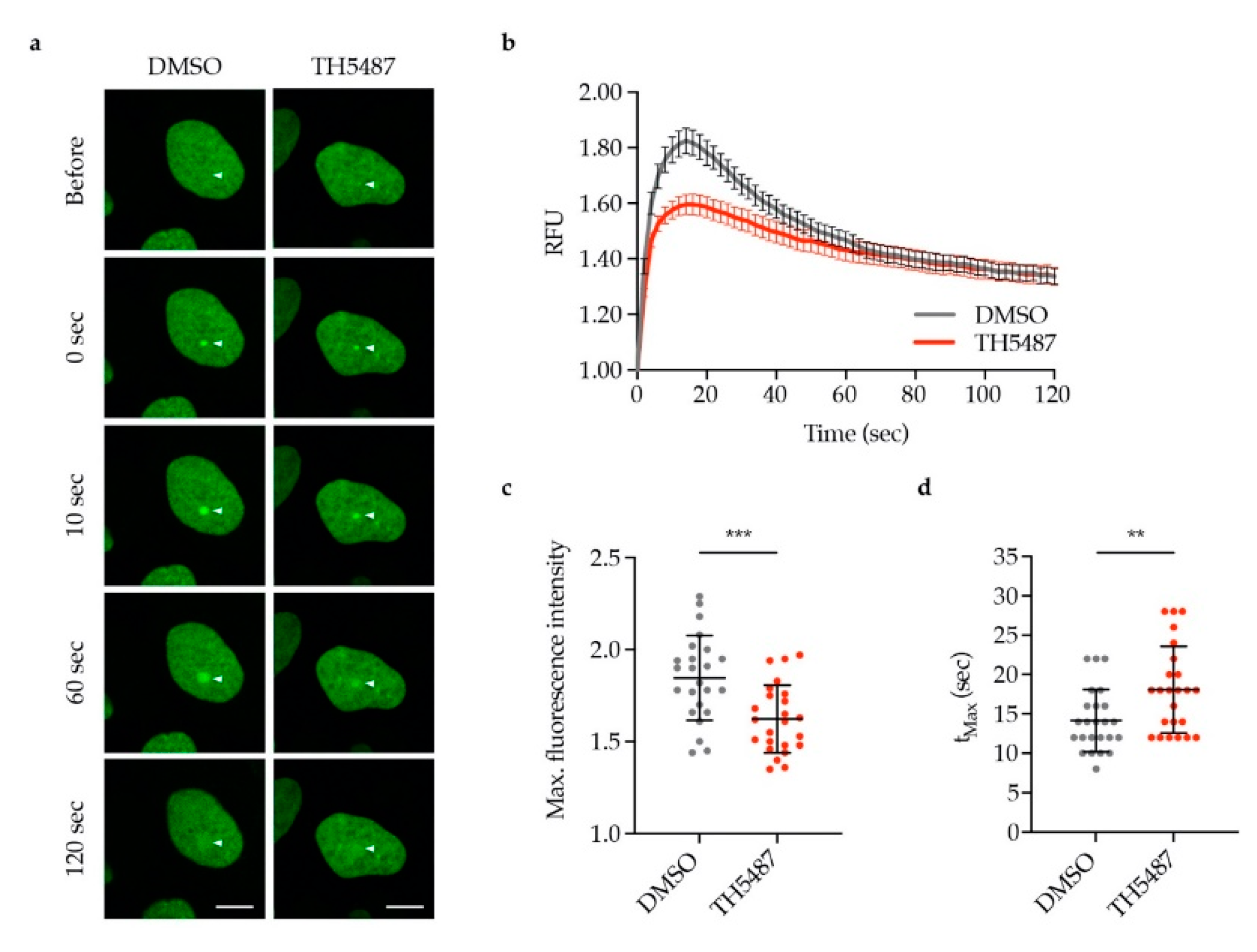
To investigate whether the reduced OGG1–chromatin binding observed in TH5487-treated cells was caused by lower recruitment or faster dissociation of OGG1, we monitored OGG1-GFP recruitment to laser-induced DNA damage sites. OGG1-GFP recruitment kinetics were found to be impaired in TH5487-treated cells exemplified by recruitment of less OGG1-GFP (Figure 4a–c) and the longer time needed for the fluorescence intensity to reach its maximum level (Figure 4d).
3.4. TH5487 Impairs OGG1 Incision and Generation of DNA Breaks
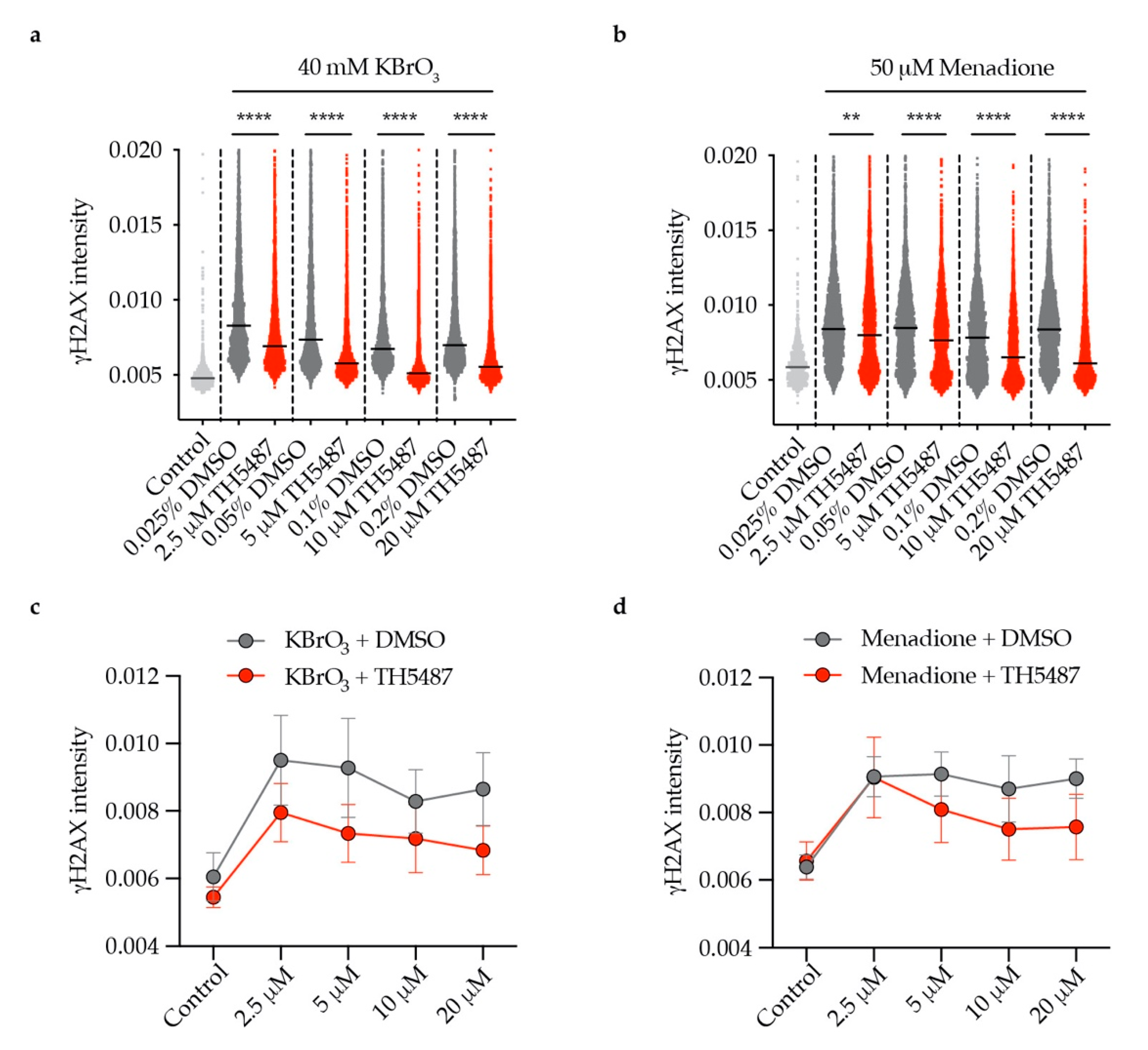
Excision of 8-oxoG lesions by OGG1 can result in the generation of DNA single-strand breaks (SSBs) [19]. Near-simultaneous attempted base excision repair (BER) of clustered oxidative lesions on opposing strands may generate double-strand breaks (DSBs) [20,21,22]. We sought to study whether TH5487 treatment impairs OGG1′s incision activity in cells, affecting DNA break generation. U2OS cells expressing OGG1-GFP were co-treated with oxidizing agents, KBrO3 or menadione and increasing concentrations of TH5487 or equivalent amount of DMSO. Both oxidizing agents have been reported to induce oxidative stress, generating 8-oxoG lesions [16,17,23]. Quantitative microcopy revealed that γH2AX formation is significantly reduced in a dose-dependent manner by TH5487 (Figure 5). This indicates that TH5487 not only prevents the binding of OGG1 to damaged DNA, but also inhibits its catalytic function in cells, leading to fewer incisions and thus fewer breaks.
4. Discussion
Although cancer cells are inherently stressed by high levels of ROS and DNA damage, DNA pathways repairing oxidative DNA damage have not been extensively targeted by inhibitors as a therapeutic approach [13]. The success of PARP inhibitors in pre-clinical and clinical studies supports the potential significance of new inhibitors for BER [24,25,26]. DNA glycosylases in particular may serve as promising anticancer targets, as mice lacking these enzymes are generally viable and fertile [27]. The high load of ROS in cancer cells and the relative abundance of guanines in GC-rich promoters and telomeres [28,29,30] have prompted us and others to develop the first small-molecule OGG1 inhibitors [14,31].
We have previously shown that TH5487 inhibits OGG1′s glycosylase activity in in vitro biochemical assays. In addition, T cell leukemia Jurkat A3 cells treated with KBrO3 and released into fresh medium containing TH5487 showed an increase in genomic 8-oxoG levels as detected by liquid chromatography–tandem mass spectrometry (LC-MS/MS) [14]. Consistently, modified comet assays demonstrated that genomic OGG1 substrates accumulate in Jurkat A3 cells treated with TH5487, as such treatment caused an increase in OGG1-induced tail length [32]. Notably, TH5487 treatment suppresses the growth of a large panel of cancer cell lines encompassing several human solid tumors including osteosarcoma, colon, glioblastoma, kidney and lung cancer but is well tolerated by non-transformed cells [32]. Here, we sought to further characterize the effect of TH5487 on cancer cells using a solid tumor cell model, U2OS. We observed that OGG1 inhibition results in 8-oxoG accumulation in DNA (Figure 1). This confirms that TH548 targets OGG1′s glycosylase activity in U2OS cells and that the previously described elevation in 8-oxoG levels upon TH5487 treatment [14] is not restricted to Jurkat A3 T lymphocytes.
Live cell imaging and FRAP assays in U2OS cells show that upon TH5487 treatment, OGG1 is more mobile (Figure 2d–f), indicating that OGG1 binding to damaged chromatin is impaired. This affirms the observed phenotype in A3 cells, where TH5487 was found to alter OGG1–chromatin dynamics [14]. The increased mobility of OGG1 excludes any entrapment of OGG1 on damaged DNA after OGG1 inhibition. This is in agreement with an almost complete lack of detectable OGG1 retention in pre-extracted cells treated with both KBrO3 and TH5487 (Figure 3). Importantly, the observed impaired OGG1–chromatin binding, together with the previously reported in vitro electrophoretic mobility shift assay (EMSA) results, support our conclusion that TH5487 hampers OGG1 binding to damaged DNA. Besides, it complements the reported crystal structure of mouse OGG1 in complex with one analogue of TH5487 confirming targeting of OGG1′s active site [14]. The recently resolved crystal structure of human OGG1 in complex with TH5487 further confirms that TH5487 targets the active site of human OGG1. OGG1 adopts a closed conformation upon binding to TH5487, which blocks OGG1′s access to its substrate DNA lesions [32]. This explains the increased mobility of OGG1 upon TH5487 treatment (Figure 2d–f).
TH5487 interferes with OGG1 binding to damaged DNA and affects its recruitment kinetics to DNA damage sites as well (Figure 4). Hence, this strongly suggests that the impaired OGG1–chromatin binding observed in inhibitor-treated cells (Figure 2 and Figure 3) is not a result of faster OGG1 dissociation from DNA. This provides an additional explanation as to why more 8-oxoG lesions are detected in oxidatively stressed cells upon treatment with TH5487 (Figure 1).
During BER of clustered oxidative DNA lesions existing on opposing strands, double-strand breaks can be introduced [20,21,22]. Our observation that the level of γH2AX is reduced in TH5487-treated cells (Figure 5) illustrates that the compound inhibits OGG1′s incision activity, verifying the reported effect of TH5487 on OGG1′s enzymatic activity in in vitro biochemical assays [14]. This inhibition is translated into accumulation of genomic 8-oxoG lesions as shown in Figure 1. A definitive quantitative method for 8-oxoG is lacking. Hence, the absolute level that cancer or normal cells can withstand is not yet clear. Further investigation is needed to avoid on-target toxicity in normal tissues.
As TH5487 selectively induces proliferation arrest in cancer cell lines but not in non-transformed cells [32], OGG1 serves as a potential target for cancer treatment. More research should be directed towards optimizing the compound’s pharmacokinetic profile to determine its efficacy in in vivo cancer models.
5. Conclusions
This study validates TH5487 as a potent OGG1 inhibitor that prevents the repair of 8-oxoG lesions and alters both OGG1–chromatin dynamics and OGG1′s recruitment kinetics to DNA damage sites. We demonstrate that TH5487 can be used to pharmacologically inhibit the glycosylase activity of OGG1, enabling future studies of OGG1′s role in different disease models.
Author Contributions
Conceptualization, B.M.F.H., O.M., and T.H.; methodology, B.M.F.H. and O.M.; data analysis, B.M.F.H. and O.M.; writing—original draft preparation, B.M.F.H.; writing—review and editing, O.M. and T.H.; supervision, T.H.; funding acquisition, T.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the European Union’s Horizon 2020 research and innovation program SYNTRAIN under the Marie Sklodowska-Curie grant agreement No. 722729, The European Research Council (ERC-AdG-695376 and TAROX Programme), Vinnova (2018-03232), Novo Nordisk (29972), The Torsten and Ragnar Söderberg Foundation, Barncancerfonden (BCF PR2018-0095 2019-2021), The Swedish Cancer Society (CAN2018/0658), and The Swedish Research Council (2015-00162).
Acknowledgments
We are grateful to T. Visnes, H. Gad, and P. Herr for sharing the expression vectors. We thank L. Sjöholm and M.K. Magnusson for administrative and technical support.
Conflicts of Interest
T.H. is listed as an inventor on a provisional U.S. patent application No. 62/636983, covering OGG1 inhibitors. The patent is fully owned by a nonprofit public foundation, the Helleday Foundation. T.H. is also a member of the foundation board developing OGG1 inhibitors for the clinic. The remaining authors declare no competing financial interests. The funders had no role in the design of the study; in the collection, analysis or interpretation of data; in the writing of the manuscript or in the decision to publish the results.
References
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R.R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free Radicals and Other Reactive Species in Disease. eLS 2015, 5, 1–9. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Santos, A.L.; Sinha, S.; Lindner, A.B. The Good, the Bad, and the Ugly of ROS: New Insights on Aging and Aging-Related Diseases from Eukaryotic and Prokaryotic Model Organisms. Oxid. Med. Cell. Longev. 2018, 2018, 1941285. [Google Scholar] [CrossRef]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative Stress and Cell Signalling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic. Biol. Med. 2017, 107, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Neeley, W.L.; Essigmann, J.M. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem. Res. Toxicol. 2006, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Visnes, T.; Grube, M.; Hanna, B.M.F.; Benitez-Buelga, C.; Cázares-Körner, A.; Helleday, T. Targeting BER enzymes in cancer therapy. DNA Repair (Amst) 2018, 71, 118–126. [Google Scholar] [CrossRef]
- Visnes, T.; Cázares-Körner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Mortusewicz, O.; Ma, H.T.; Herr, P.; Poon, R.R.Y.; Helleday, T.; Qian, C. Timeless Interacts with PARP-1 to Promote Homologous Recombination Repair. Mol. Cell 2015, 60, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Kasai, H.; Nishimura, S.; Kurokawa, Y.; Hayashi, Y. Oral administration of the renal carcinogen, potassium bromate, specifically produces 8-hydroxydeoxyguanosine in rat target organ dna. Carcinogenesis 1987, 8, 1959–1961. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L. The role of glutathione in DNA damage by potassium bromate in vitro. Mutagenesis 2000, 15, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amouroux, R.; Campalans, A.; Epe, B.; Radicella, J.P. Oxidative stress triggers the preferential assembly of base excision repair complexes on open chromatin regions. Nucleic Acids Res. 2010, 38, 2878–2890. [Google Scholar] [CrossRef] [Green Version]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Galick, H.; Wallace, S.S. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst) 2004, 3, 1323–1334. [Google Scholar] [CrossRef]
- Yang, N.; Chaudhry, M.A.; Wallace, S.S. Base excision repair by hNTH1 and hOGG1: A two edged sword in the processing of DNA damage in γ-irradiated human cells. DNA Repair (Amst) 2006, 5, 43–51. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-strand DNA Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, S.; Ohno, M.; Tsuchimoto, D.; Sakumi, K.; Furuichi, M.; Nakabeppu, Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008, 27, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Meira, L.B. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage Version 7. DNA Repair (Amst) 2006, 5, 189–209. [Google Scholar] [CrossRef]
- Ding, Y.; Fleming, A.M.; Burrows, C.J. Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-dihydroguanine by OG-Seq. J. Am. Chem. Soc. 2017, 139, 2569–2572. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor κB-driven gene expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef] [Green Version]
- Amente, S.; Di Palo, G.; Scala, G.; Castrignanò, T.; Gorini, F.; Cocozza, S.; Moresano, A.; Pucci, P.; Ma, B.; Stepanov, I.; et al. Genome-wide mapping of 8-oxo-7,8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019, 47, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Tahara, Y.K.; Auld, D.; Ji, D.; Beharry, A.A.; Kietrys, A.M.; Wilson, D.L.; Jimenez, M.; King, D.; Nguyen, Z.; Kool, E.T. Potent and Selective Inhibitors of 8-Oxoguanine DNA Glycosylase. J. Am. Chem. Soc. 2018, 140, 2105–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visnes, T.; Benítez-Buelga, C.; Cázares-Körner, A.; Sanjiv, K.; Hanna, B.M.F.; Mortusewicz, O.; Rajagopal, V.; Albers, J.J.; Hagey, D.W.; Bekkhus, T.; et al. Targeting OGG1 arrests cancer cell proliferation by inducing replication stress. Nucleic Acids Res. 2020. Accepted. [Google Scholar]
Figure 1.
OGG1 inhibition with TH5487 results in accumulation of genomic 8-oxoG base lesions. (a) Scheme of TH5487 treatment to study the repair kinetics of genomic 8-oxoG. (b) Fluorescence signal intensity of 8-oxoG at the indicated time points. U2OS cells were pre-challenged with 20 mM KBrO3 for 1 h, then allowed to recover in a medium containing either 0.1% DMSO or 10 μM TH5487 for the indicated time points. Scatterplot shows data of at least 500 cells for each treatment condition with the mean from three independent experiments. Statistical significance was determined using an unpaired two-sided t-test (** P < 0.01, **** P < 0.0001). (c) Representative confocal images of U2OS cells showing accumulation of genomic 8-oxoG lesions. Scale bar: 50 μm.
Figure 1.
OGG1 inhibition with TH5487 results in accumulation of genomic 8-oxoG base lesions. (a) Scheme of TH5487 treatment to study the repair kinetics of genomic 8-oxoG. (b) Fluorescence signal intensity of 8-oxoG at the indicated time points. U2OS cells were pre-challenged with 20 mM KBrO3 for 1 h, then allowed to recover in a medium containing either 0.1% DMSO or 10 μM TH5487 for the indicated time points. Scatterplot shows data of at least 500 cells for each treatment condition with the mean from three independent experiments. Statistical significance was determined using an unpaired two-sided t-test (** P < 0.01, **** P < 0.0001). (c) Representative confocal images of U2OS cells showing accumulation of genomic 8-oxoG lesions. Scale bar: 50 μm.

Figure 2.
TH5487 alters OGG1–chromatin dynamics. (a) Representative false color images of nontreated and KBrO3-treated U2OS cells expressing OGG1-GFP are shown. Dashed outlines indicate bleached areas. Scale bar: 5 μm. (b) Quantification of fluorescence recovery after photobleaching (FRAP). U2OS cells expressing OGG1-GFP were treated or not with 40 mM KBrO3 for 1 h, a nuclear region was bleached, and recovery of fluorescence after photobleaching was recorded. RFU (relative fluorescence units). (c) Quantification of OGG1’s mobile fraction. Exposure to 40 mM KBrO3 for 1 h reduced the nuclear mobility of OGG1-GFP. (d) Representative false color images of DMSO-treated and TH5487-treated U2OS cells expressing OGG1-GFP are shown. Dashed outlines indicate bleached areas. Scale bar: 5 μm. (e) Quantification of FRAP experiments where U2OS cells expressing OGG1-GFP were treated with 40 mM KBrO3 for 1 h, followed by another hour of additional exposure to 10 μM TH5487 or 0.1% DMSO. (f) Quantification of OGG1’s mobile fraction. Treatment with TH5487 resulted in higher nuclear mobility of OGG1-GFP. Data are means ± SEM (b,e) or means ± SD (c,f) of three independent experiments. At least 35 cells from three independent experiments were quantified. Statistical significance was determined using unpaired two-sided t-tests (**** P < 0.0001).
Figure 2.
TH5487 alters OGG1–chromatin dynamics. (a) Representative false color images of nontreated and KBrO3-treated U2OS cells expressing OGG1-GFP are shown. Dashed outlines indicate bleached areas. Scale bar: 5 μm. (b) Quantification of fluorescence recovery after photobleaching (FRAP). U2OS cells expressing OGG1-GFP were treated or not with 40 mM KBrO3 for 1 h, a nuclear region was bleached, and recovery of fluorescence after photobleaching was recorded. RFU (relative fluorescence units). (c) Quantification of OGG1’s mobile fraction. Exposure to 40 mM KBrO3 for 1 h reduced the nuclear mobility of OGG1-GFP. (d) Representative false color images of DMSO-treated and TH5487-treated U2OS cells expressing OGG1-GFP are shown. Dashed outlines indicate bleached areas. Scale bar: 5 μm. (e) Quantification of FRAP experiments where U2OS cells expressing OGG1-GFP were treated with 40 mM KBrO3 for 1 h, followed by another hour of additional exposure to 10 μM TH5487 or 0.1% DMSO. (f) Quantification of OGG1’s mobile fraction. Treatment with TH5487 resulted in higher nuclear mobility of OGG1-GFP. Data are means ± SEM (b,e) or means ± SD (c,f) of three independent experiments. At least 35 cells from three independent experiments were quantified. Statistical significance was determined using unpaired two-sided t-tests (**** P < 0.0001).

Figure 3.
TH5487 treatment affects OGG1 retention in cells exposed to oxidative stress. (a) OGG1–chromatin binding. Representative confocal images of U2OS cells after in situ extraction. Scale bar: 20 μm. (b) Fluorescence signal intensity of retained OGG1-GFP. U2OS cells were exposed to the indicated treatment conditions, then in situ extraction with 0.1% Triton for 1 min was performed prior to fixation with 4% paraformaldehyde. Scatterplot shows OGG1-GFP nuclear fluorescence signal intensity of at least 1300 cells for each treatment condition and mean fluorescence intensity ± SD of three independent experiments. Statistical significance was determined using unpaired two-sided t-test (**** P < 0.0001).
Figure 3.
TH5487 treatment affects OGG1 retention in cells exposed to oxidative stress. (a) OGG1–chromatin binding. Representative confocal images of U2OS cells after in situ extraction. Scale bar: 20 μm. (b) Fluorescence signal intensity of retained OGG1-GFP. U2OS cells were exposed to the indicated treatment conditions, then in situ extraction with 0.1% Triton for 1 min was performed prior to fixation with 4% paraformaldehyde. Scatterplot shows OGG1-GFP nuclear fluorescence signal intensity of at least 1300 cells for each treatment condition and mean fluorescence intensity ± SD of three independent experiments. Statistical significance was determined using unpaired two-sided t-test (**** P < 0.0001).

Figure 4.
TH5487 treatment reduces OGG1 recruitment to laser-induced DNA damage sites. (a) Representative images of DMSO- and TH5487-treated U2OS cells expressing OGG1-GFP. Arrows indicate the point of laser irradiation. Scale bar: 10 μm. (b) OGG1 recruitment kinetics. U2OS cells expressing OGG1-GFP were pre-sensitized with Hoechst and treated with either 0.1% DMSO or 10 μM TH5487 for 1 h, then DNA damage was induced with a 405 nm laser. RFU (relative fluorescence units). (c) Quantification of the maximum fluorescence intensity upon OGG1-GFP recruitment. (d) Quantification of the time needed to reach maximum fluorescence of OGG1-GFP. The scatterplots show the data of at least 24 cells for each treatment condition. Data are means ± SEM (b) or means ± SD (c,d) of two independent experiments. Statistical significance was determined using unpaired two-sided t-tests (** P < 0.01, *** P < 0.001).
Figure 4.
TH5487 treatment reduces OGG1 recruitment to laser-induced DNA damage sites. (a) Representative images of DMSO- and TH5487-treated U2OS cells expressing OGG1-GFP. Arrows indicate the point of laser irradiation. Scale bar: 10 μm. (b) OGG1 recruitment kinetics. U2OS cells expressing OGG1-GFP were pre-sensitized with Hoechst and treated with either 0.1% DMSO or 10 μM TH5487 for 1 h, then DNA damage was induced with a 405 nm laser. RFU (relative fluorescence units). (c) Quantification of the maximum fluorescence intensity upon OGG1-GFP recruitment. (d) Quantification of the time needed to reach maximum fluorescence of OGG1-GFP. The scatterplots show the data of at least 24 cells for each treatment condition. Data are means ± SEM (b) or means ± SD (c,d) of two independent experiments. Statistical significance was determined using unpaired two-sided t-tests (** P < 0.01, *** P < 0.001).

Figure 5.
Concentration-dependent reduction of OGG1 incisions by TH5487. (a,b) Reduced nuclear γH2AX intensity in U2OS cells treated with (a) KBrO3 or (b) menadione and increasing concentrations of TH5487 for 1 h. (c,d) Mean values of (a) and (b). Scatterplots show data of at least 1000 (control) or 3300 (all other conditions) cells. Data are presented as median (a,b) and mean (c,d) ± SEM of two independent experiments with two technical repeats each. Statistical significance was determined using unpaired two-sided t-tests (** P < 0.01, **** P < 0.0001).
Figure 5.
Concentration-dependent reduction of OGG1 incisions by TH5487. (a,b) Reduced nuclear γH2AX intensity in U2OS cells treated with (a) KBrO3 or (b) menadione and increasing concentrations of TH5487 for 1 h. (c,d) Mean values of (a) and (b). Scatterplots show data of at least 1000 (control) or 3300 (all other conditions) cells. Data are presented as median (a,b) and mean (c,d) ± SEM of two independent experiments with two technical repeats each. Statistical significance was determined using unpaired two-sided t-tests (** P < 0.01, **** P < 0.0001).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hanna, B.M.F.; Helleday, T.; Mortusewicz, O. OGG1 Inhibitor TH5487 Alters OGG1 Chromatin Dynamics and Prevents Incisions. Biomolecules 2020, 10, 1483. https://doi.org/10.3390/biom10111483
AMA Style
Hanna BMF, Helleday T, Mortusewicz O. OGG1 Inhibitor TH5487 Alters OGG1 Chromatin Dynamics and Prevents Incisions. Biomolecules. 2020; 10(11):1483. https://doi.org/10.3390/biom10111483
Chicago/Turabian StyleHanna, Bishoy M. F., Thomas Helleday, and Oliver Mortusewicz. 2020. "OGG1 Inhibitor TH5487 Alters OGG1 Chromatin Dynamics and Prevents Incisions" Biomolecules 10, no. 11: 1483. https://doi.org/10.3390/biom10111483
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.